Proteins are microscopic machines involved in nearly all aspects of life. For them to work correctly, most need to fold into precise three-dimensional structures that enable their atoms to catalyze chemical reactions, bind to other molecules, and produce mechanical forces. The overarching goal of the lab is to better understand how protein structure and dynamics determine activity and ultimately enable rational manipulation for therapeutic or synthetic applications. We take a distinctly multidisciplinary approach that focuses on three interrelated areas:
Dynamics of Computationally Designed Proteins
Computational protein design is becoming an increasingly powerful tool for creating entirely new proteins for therapeutic development, biophysical characterization, and materials design. Nearly all protein design approaches aim to create proteins that fold into a single target structure. However, most designs end up populating an ensemble of conformations. We aim to characterize how the dynamics of these proteins affect their function, in the process enabling targeted manipulation of the energy landscape to favor active conformations and disfavor undesired states.
Mechanisms of mini Fluorescence Activating Protein (mFAP) excited state stabilization
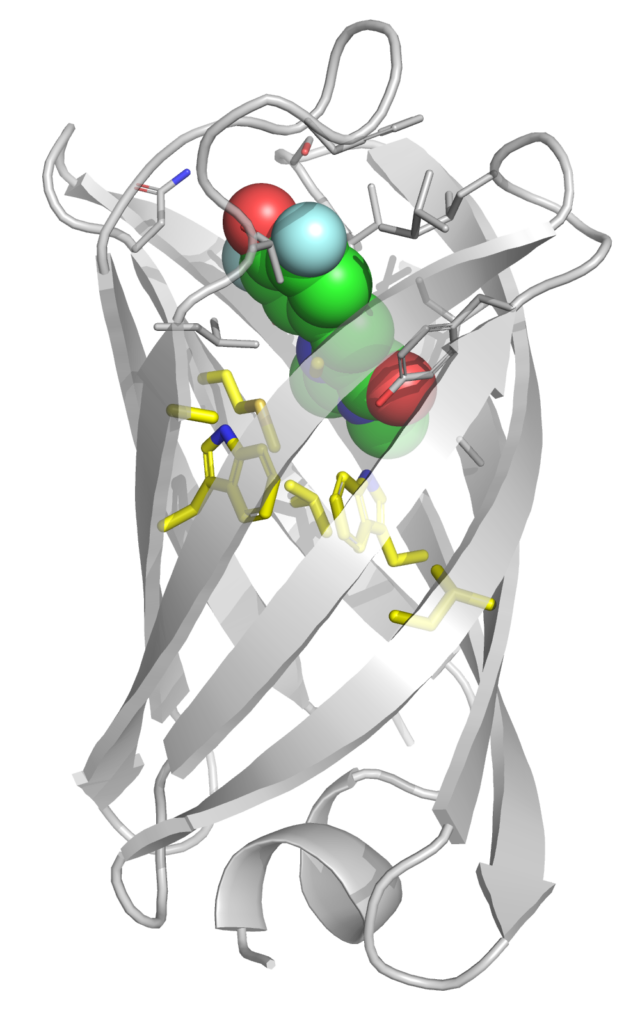
The recent creation of mini beta-barrel proteins capable of binding to and activating the florescence of a small-molecule chromophore is a prime example of the power of de novo protein design. The primary hypothesis for how mFAPs function is that they keep the chromophore ligand in a relatively rigid planar conformation long enough to fluoresce. While rational optimization of folding and binding are relatively mature disciplines, approaches for increasing biomolecular rigidity are less established. We have developed a computational protocol to predict mFAP rigidity/fluorescence and are using this as a model system to understand how proteins stabilize small-molecules in functional conformations. People: Meagan MacDonald, Roberta Akrong, Gavin Brangham, Farah Hasanain, Amari Stuppard (collaboration with Prof. Carlos Jimenez-Hoyos and Jeff Keyes)
Dynamics of de novo designed mini proteins and engineering away undesired conformations
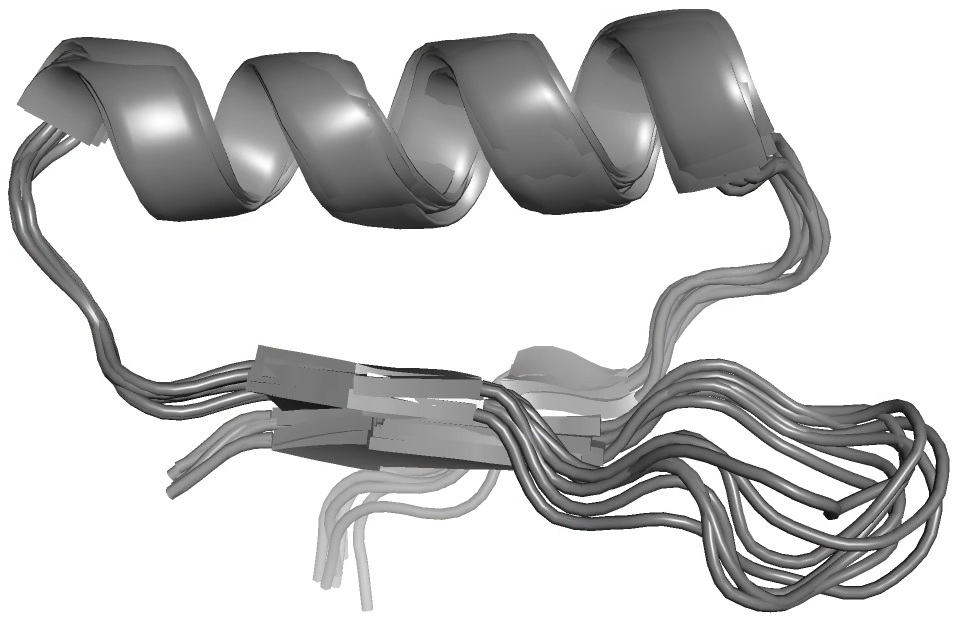
High throughput DNA synthesis and library screening now enable the computational design and testing of thousands of mini proteins at once. This technology has been recently used to rapidly develop inhibitors of botulinum toxin, influenza, and SARS-CoV-2. The resulting proteins are often hyper-stable, reducing or eliminating the need for refrigerated storage, and can be administered by nasal spray. While this new class of biomolecules shows great promise for use as drugs, we have found that they can take on undesired alternate conformations, which could be deleterious to their intended functions. We are currently characterizing these alternate states and redesigning the proteins to avoid them. We are also exploring how their dynamics varies over the large temperature range in which they are stable. People: Kat Blejec, Owen Rogers, Benjamin Levine, William Wise, Alex Kermath, Tihitina Gebeyehu
Developing Methods for Computational Modeling
Much of our work involves computational simulation and modeling, which provides an extraordinary amount of detail. We also make use of high-resolution biophysical techniques like nuclear magnetic resonance (NMR) to accurately determine both structure and dynamics at the atomic level. To enable these investigations, we develop new computational techniques to better harness experimental NMR data for simulation and modeling.
Quantifying protein motion from NOE data using the Kinetic Ensemble approach
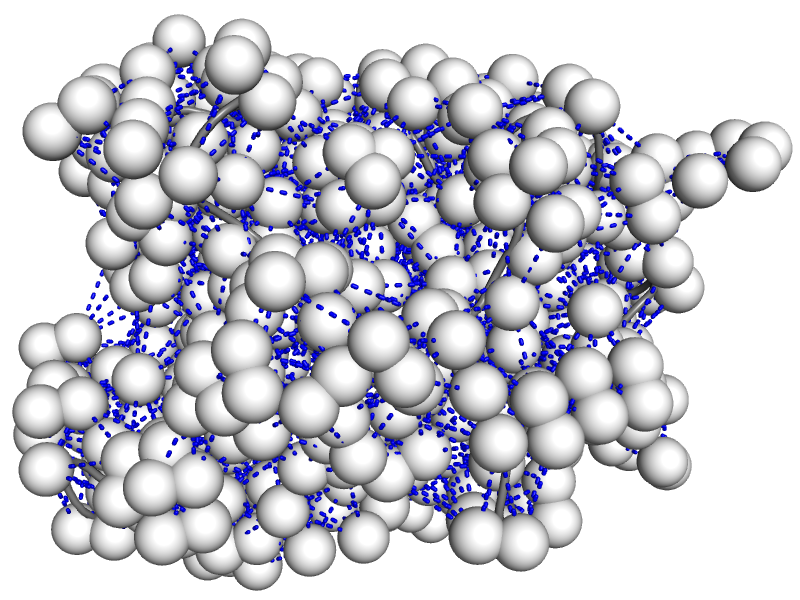
The nuclear Overhauser effect (NOE) is the basis for most protein structures solved via NMR. However, during structure determination, NOE data is often used in a very qualitative manner with large uncertainties. Furthermore, critical aspects of protein motion are almost entirely neglected with current structure refinement approaches. We have developed the Kinetic Ensemble approach that allows the fully quantitative modeling of NOE data from existing ensembles of multiple structures. We are now in the process of adapting the approach to enable much more accurate structure determination. People: Amr Alhossary, Abdiasis Daauud, Shikhar Gupta
Fitting NMR spectra to extract hidden information about protein structure and dynamics
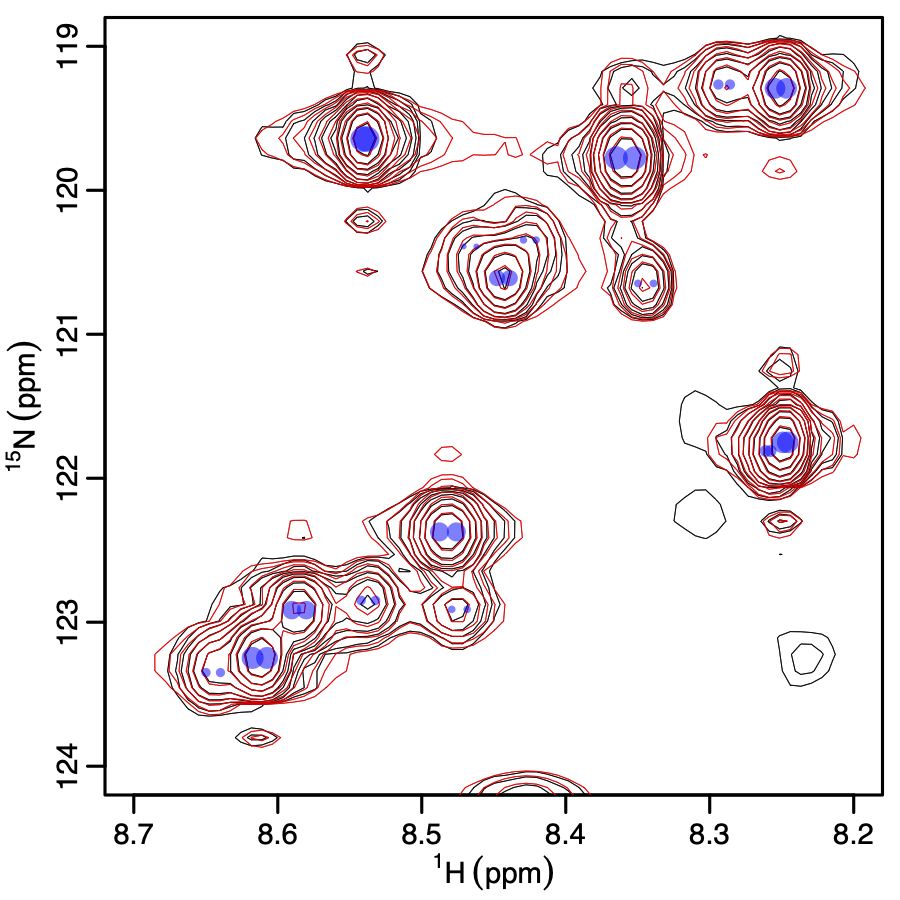
NMR spectra capture a plethora of information about molecular structure. However, there are often crowded regions where isolation of peaks is difficult, complicating the structural interpretation. In addition, the peak shapes contain useful information about dynamics and couplings between nearby atoms. To address problems of overlap and accurately quantifying valuable information from peak shapes, we have developed FitNMR. We continue to develop it and other methods to extract previously unusable structural data from NMR spectra. People: Kat Blejec, Abdiasis Daauud
Reshaping free energy landscapes using computational protein design
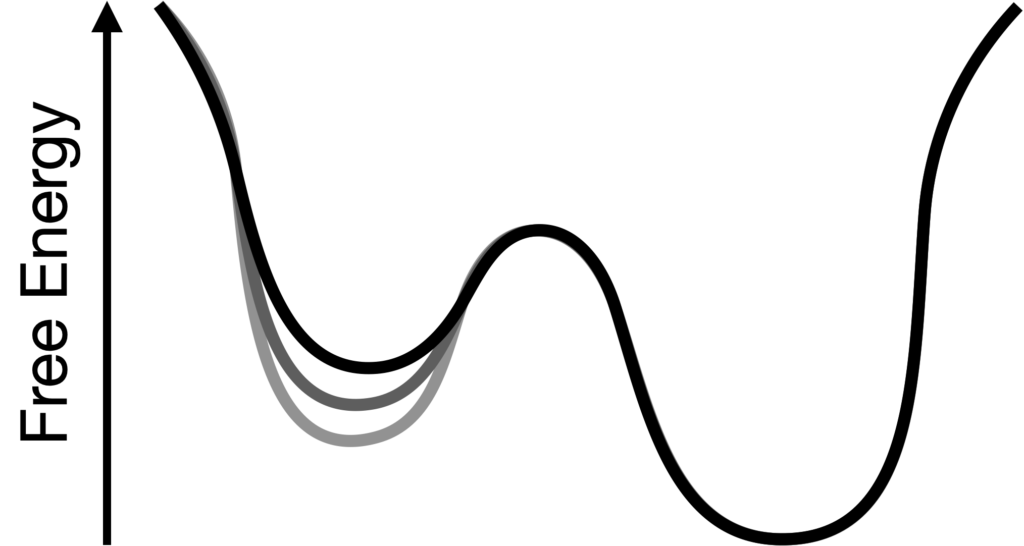
Proteins are in constant motion and interconvert between different conformational states. The function of proteins often depends on them taking on very particular structures or avoiding aberrant conformations which may have toxic effects. We are using a combination of molecular dynamics simulations and protein design algorithms to develop generalizable methods that rationally alter the free energies of both favored and disfavored states, allowing optimization of many kinds of protein functions. People: Owen Rogers, Gavin Brangham, Chukwudi Udechukwu
Mechanisms of Allosterically Acting Mutations in Natural Proteins
Traditional efforts to understand how many processes work have focused on the area of the protein immediately around where it interacts with other molecules (sometimes called the “first shell” of the active site or binding site). Several of our projects aim to determine the atomic-level mechanisms of how changes in the so-called “second shell” and beyond propagate through the protein and ultimately affect function. This can enable protein activity to be regulated by distant events through a process called allostery. Furthermore, deleterious mutations far removed from the active site make the protein malfunction and cause disease. The common mechanistic question about these examples is the following: how do atomic rearrangements propagate from one part of the structure to another? Even more intriguingly, how can a signal propagate without distinct structural changes?
Mutational effects on the maturation of ALS-related Superoxide Dismutase 1 (SOD1)
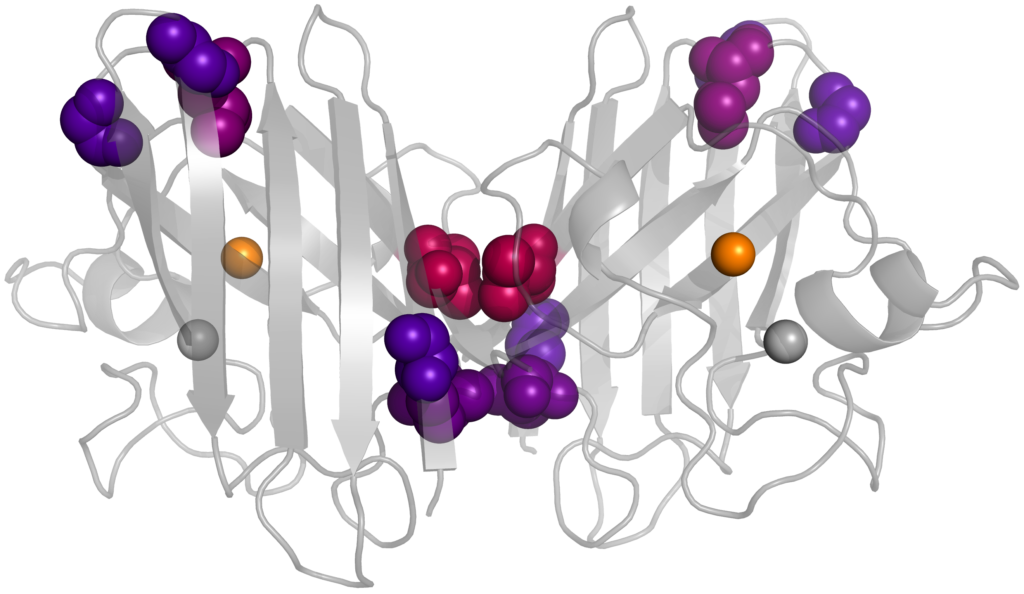
Amyotrophic Lateral Sclerosis (ALS), otherwise known as Lou Gehrig’s disease, affects approximately 6 people in 100,000 annually. It affects motor neurons, gradually leading to a loss of muscle control and often death. While SOD1 was the earliest protein linked to the disease nearly 30 years ago, the mechanism by which it causes ALS is still unknown. An emerging hypothesis is that non-native interactions involving immature forms of the protein are disease-causing. We are using alchemical free energy simulations to study how several ALS-associated mutations affect SOD1 maturation. These alchemical methods morph the chemical structure of a protein side chain from one amino acid type to another during a simulation, which would be impossible outside of a computer program. Application of several thermodynamic techniques then enables the calculation of how mutation perturbs the SOD1 free energy landscape, with the goal of helping to uncover the disease mechanism and eventually develop treatments. People: Owen Rogers (collaboration with Prof. Alison O’Neil)
Xylanase A loop dynamics and catalysis
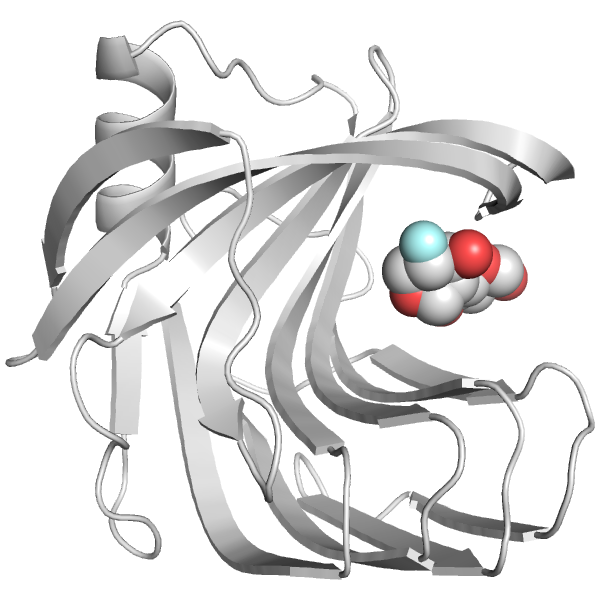
Xylanase A is a bacterial enzyme that breaks down xylan, which forms a major part the plant cell wall and comprises 20-35% of terrestrial biomass. Xylan is a potential feedstock for second-generation biofuels, making characterization and engineering of xylan-processing enzymes important for biofuel development. We are studying a set of mutations that we have found to both increase the catalytic rate and be more efficient at lower substrate concentrations. We are investigating the substrate dependence of this catalytic enhancement, and how the mutations reshape the energy landscape of the protein to favor a rarely observed alternate conformation. People: Meagan MacDonald